
Legal framework: from Directives to Regulations
The EU legal framework on the safety and performance of medical devices was established in the 1990s with three directives intended to harmonize the laws relating to medical devices and in vitro diagnostics within the European Union.
- 1990 AIMDD – Directive on active implantable medical devices (Council Directive 90/385/EEC)
- 1993 MDD – Medical Devices Directive(Council Directive 93/42/EEC)
- 1998 IVDD – In Vitro Diagnostics Directive (Directive 98/79/EC)
These directives set out the goals that EU countries should achieve but allowed for a certain flexibility in implementation. This flexibility and differences in transposition of directives to national legislation led to variations in how the European rules were applied across different countries, creating inconsistencies in the regulatory landscape.
Already in the early 2000s, the EU started revising the laws governing medical devices and in vitro diagnostics to align with the developments of the sector and to provide a legal answer to concerns about the adequacy of the existing framework. The priority was to adapt the legislation to ensure a robust, transparent and sustainable regulatory framework and maintain a high level of safety, while supporting innovation.
Two new regulations on medical devices (MDR) and in vitro diagnostic medical devices (IVDR) were approved in May 2017:
- MDR: Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices, replaced the AIMDD and MDD directives on May 26, 2021. 1
- IVDR: Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices replacing IVDD directive 98/79/EC as of May 26, 2022. 2
Unlike directives, regulations are directly applicable in all EU member countries without the need for transposition into national legislation. This ensures a more uniform application of the rules across all EU countries, enhancing the overall consistency and transparency of the regulatory framework.
In addition to taking over the provisions of the Directives , the medical devices and in vitro diagnostics Regulations contain a number of additional and enhanced provisions:
- Expanded Definition of Medical Devices: The MDR broadens the definition of a medical device to include products for cleaning, disinfection, or sterilization of devices, whereas the IVDR now also includes software only devices.
- Stricter risk classification: with more devices classified in higher risk categories for medical devices, and transition from a list-based to a rule-based device classification, with more devices requiring oversight of Notified Bodies for in vitro diagnostics.
- Enhanced Post-Market Surveillance: The MDR imposes more stringent post-market surveillance requirements, ensuring ongoing compliance and safety monitoring throughout the device’s lifecycle.
- Introduction of UDI System: Unique Device Identification (UDI) system to enhance traceability of devices and improve vigilance. All devices on the EU market will in term be centralized in the EUDAMED database.
- Person responsible for Regulatory Compliance: The MDR requires manufacturers to designate a person responsible for regulatory compliance, ensuring a specific point of accountability within the organization.
- More rigorous Clinical Evaluation: The MDR has more rigorous clinical evaluation requirements, necessitating robust clinical data to support the safety and performance claims of the device.
The additional regulatory requirements imposed on both manufacturers and notified bodies created substantial certification bottlenecks. To prevent shortages on the market that could deprive patients of essential medical devices, the original transition period of the MDR has been extended twice, the latest amendment (Regulation (EU) 2023/607) 3 conditionally extending the transition period up to December 2028 at the latest. On April 25, 2024, the European Parliament voted to conditionally extend the IVDR transition period, based on device risk class, to December 2029 at the latest, but this extension awaits issuance of an amending regulation to come into effect.
Systematic Literature Reviews under the MDR and IVDR Regulations
A thorough and systematic analysis of existing evidence under the form of a systematic literature review is required for both medical devices and in vitro diagnostics. Both regulations similarly abstain from prescribing specific requirements for such literature reviews, other than that they must be thorough, comprehensive and report on both favorable and unfavorable data. In both cases the literature review is important to establish the state-of-the-art. The key difference between medical devices and IVDs in this case concerns the purpose and the place of systematic literature reviews in relation to other regulatory documentation.
For medical devices, the systematic literature review is a cornerstone of the clinical evaluation process which requires manufacturers to provide clinical evidence demonstrating the safety and performance of medical devices. Annex XIV part A for the MDR prescribes systematic review of the literature to identify clinical data related to the device and identify any gaps in the existing clinical evidence.
For in vitro diagnostic devices, on the other hand, the systematic literature review is important for alignment with the general safety and performance requirements as described in Annex I of the IVDR. Further, the systematic literature review is a key part of the performance evaluation of IVDs described in IVDR Annex XIII.
Therefore, clinical evidence for medical devices may include clinical investigations and data from post-market surveillance (PMS), whereas the focus for IVD may be more on analytical and clinical performance studies, rather than the traditional clinical studies.
Guidance Documents for Systematic Literature Reviews
MEDDEV
Under the framework of the Directives, The European Commission developed the MEDDEV guidelines to help manufacturers, notified bodies, and other stakeholders understand and comply with the requirements of the MDD more effectively. These guidelines covered a wide range of topics related to medical devices, including clinical evaluations, risk management, post-market surveillance, software validation, and conformity assessment procedures. They provided detailed explanations, examples, and recommendations to support the implementation of MDD requirements in practical terms.
MEDDEV 2.7/1 Revision 4 provides guidance on clinical evaluation for medical devices, including the use of clinical data derived from literature. 4
Whereas the regulations governing clinical evaluation have changed between directives and regulations, the general principles of carrying out a clinical evaluation have not. And for now, MEDDEV 2.7/1 Rev. 4 is still one of the go-to documents for clinical evaluation. The sections of MEDDEV 2.7/1 Revision 4 that remain relevant to the MDR are listed in MDCG 2020-6.
MDCG (Medical Device Coordination Group)
In the context of the regulations, the MEDDEV guidance documents have been and are still being updated into MDCG guidance. Ongoing and planned MDCG guidance can be consulted here.
The following MDCG Guidelines have been published impacting clinical evaluation assessments.
- MDCG 2020-5 covers the demonstration of equivalence, based on data pertaining to an already existing device on the market, for the purpose of CE-marking under the MDR. 5
- MDCG 2020-6 provides guidance for manufacturers and notified bodies on how to collect and analyze clinical data related to these legacy devices. 6
- MDCG 2020-13 is aimed at providing guidance on the clinical evaluation process for medical devices. Given the complexity and significance of clinical data in proving the safety and performance of a device, MDCG 2020-13 acts as a crucial resource. 7
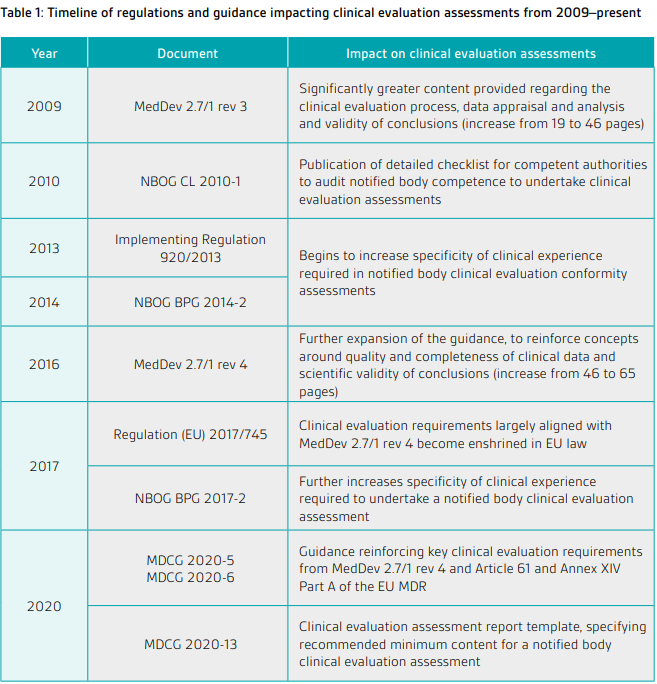
Table from BSI
- MDCG 2022-2 on performance evaluation of in vitro diagnostics requires literature searches to be systematic, conducted according to defined search protocol and properly documented, so that methods can be appraised, results verified and searches are reproducible. 8
The MDCG documents in general do not currently offer any specific concrete guidance on how the literature search should be carried out. MDCG 2020-6 however does include two appendices that will be helpful in planning and carrying out a clinical evaluation for a legacy medical device:
- Appendix 1 outlines which sections of MEDDEV 2.7/1 Rev. 4—which offers guidance on the general process for conducting a clinical evaluation—are still relevant for legacy devices under MDR.
- Appendix 2 lays out the minimum elements that a modified clinical evaluation plan for legacy devices should include. The MDCG document 2020-13 “Clinical evaluation assessment report template” might also be useful. It is primarily aimed at clinical evaluation reviewers, particularly notified bodies, but it also provides indirect guidance for anyone carrying out a clinical evaluation. Section D deals with literature search and literature review. The requirements listed in this section are the same as the ones in MEDDEV 2.7/1 Revision 4.
The focus is on:
- Search categories (e.g. device search or state of the art including clinical condition)
- Scope of the search strategy
- Search and review methods
- Literature search documentation
IMDRF (International Medical Device Regulators Forum)
Enjoyed this piece?
References and Reading
References and Reading
- European Parliament, Council of the European Union. Regulation (EU) 2017/745 of the European Parliament and of the Council of 5 April 2017 on medical devices. Off J Eur Union. 2017;10(December 2016):1-21. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=CELEX%3A32017R0745.
- European Parliament, Council of the European Union. Regulation (EU) 2017/746 of the European Parliament and of the Council of 5 April 2017 on in vitro diagnostic medical devices. Off J Eur Union. 2017;60(L117):2-175. https://eur-lex.europa.eu/eli/reg/2017/746/oj.
- Parliament E, Council of the European Union. REGULATION (EU) 2023/607. Off J Eur Union. 2023;66(L80):24. https://eur-lex.europa.eu/legal-content/EN/TXT/?uri=uriserv%3AOJ.L_.2023.080.01.0024.01.ENG&toc=OJ%3AL%3A2023%3A080%3ATOC.
- European Commission. MEDDEV. 2.7.1 Rev.4: Clinical Evaluation: A Guide for Manufacturers And Notified Bodies Under Directives 93/42/EEC and 90/385/EEC. MEDDEV 271 Rev4. 2009;(April 2003):1-65.
- Medical Devices Coordination Group (MDCG). MDCG 2020-5_Clinical Evaluation – Equivalence. 2020:1-20. https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2020_5_guidance_clinical_evaluation_equivalence_en_0.pdf.
- Medical Devices Coordination Group (MDCG). MDCG 2020-6 Clinical evidence needed for medical devices previously CE marked under Directives 93/42/EEC or 90/385/EEC. 2020:1-22. https://health.ec.europa.eu/system/files/2020-09/md_mdcg_2020_6_guidance_sufficient_clinical_evidence_en_0.pdf.
- Medical Devices Coordination Group (MDCG). MDCG 2020-13 Clinical Evaluation Assessment Report Template.; 2020.
- Medical Device Coordination Group. MDCG 2022-2 Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs). 2022:1-31. https://ec.europa.eu/health/system/files/2022-01/mdcg_2022-2_en.pdf.
- International Medical Device Regulators Forum. Clinical Evaluation (Imdrf Mdce Wg/N56Final:2019).; 2019. https://www.imdrf.org/documents/clinical-evaluation.



